The biological mechanisms that give rise to the cognitive decline of Alzheimer's disease could be due for a major rethink, according to new research.
It's long been thought that the neurodegeneration of Alzheimer's is caused by beta-amyloid plaques – sticky congregations of a protein called amyloid precursor protein (APP), which break down into fragments and clump together into misfolded, toxic aggregates in the brain, impeding neural communication.
The idea is these beta-amyloid plaques are what's responsible for neuron death in cases of Alzheimer's disease – either directly, or by giving rise to tau phosphorylation, in which the protein tau is bent into neurofibrillary tangles that disrupt nutrient supply to brain cells, eventually killing them.
But new findings by researchers at the University of Queensland in Australia suggest some of these assumptions may be flawed.
"Our data challenges the current dogma in the field that amyloid plaques are sufficient to cause neurodegenerative changes associated with Alzheimer's disease," explains one of the team, stem cell biologist Ernst Wolvetang from the Australian Institute for Bioengineering and Nanotechnology.
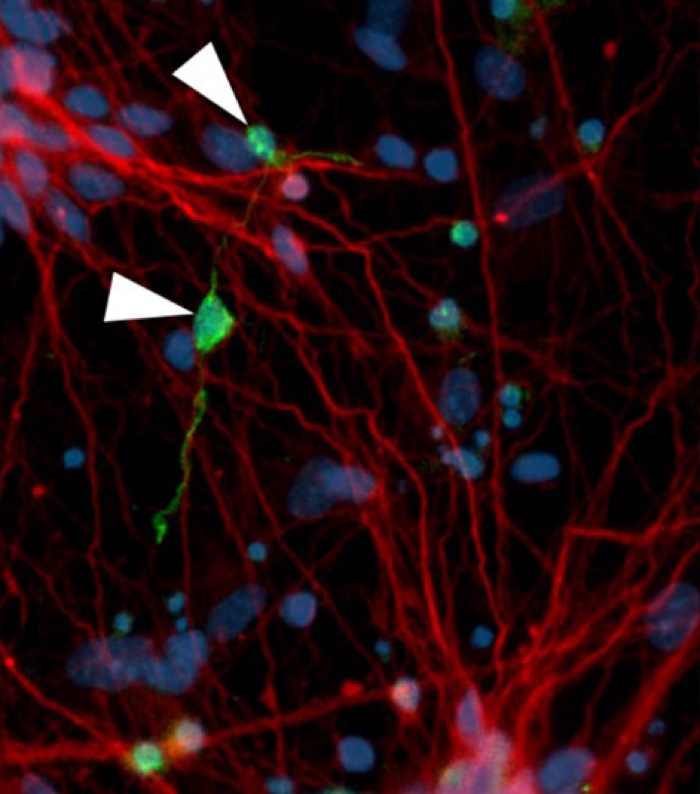 Neurofibrillary tangle-like structures (green) in affected neurons (red) (D Ovchinnikov)
Neurofibrillary tangle-like structures (green) in affected neurons (red) (D Ovchinnikov)
To investigate these mechanisms, Wolvetang and fellow researchers used stem cells from people with Down syndrome, who end up with an extra copy of the amyloid precursor protein (APP) gene, due to having an extra copy of chromosome 21.
This increased dosage of APP is what's thought to be responsible for people with Down syndrome commonly developing Alzheimer's disease.
But when the team grew the stem cells into neurons in vitro and used CRISPR gene editing to manipulate the APP to normal levels, they observed no changes in tau phosphorylation took place.
While the experiments confirmed increased levels of APP do lead to increased beta-amyloid plaques, strangely, this in itself doesn't seem to cause an increase in neuronal cell death, or in tau's toxic neurofibrillary tangles.
"It suggests that beta-amyloid may not be the central driver of AD-associated neural cell death, and is not directly responsible for tau pathology (at least in our model)," Wolvetang explained to ScienceAlert.
"I would like to stress that we are careful not to over-interpret our data. Nevertheless decades of research and billions of dollars have been expended on amyloid-based therapeutics, and to date these have largely failed.
"Our data add to an increasing number of studies that indicate that this hypothesis perhaps needs to be reevaluated."
But if levels of beta-amyloid and APP aren't directly related to tau tangling – and the cognitive decline that results – what is causing neural death in Alzheimer's disease?
The researchers don't know for sure, but they suggest we need to focus more specifically on tau processes and pathology, given amyloids may not be as involved as we once thought.
"In the Down syndrome context, we are looking at other chromosome 21 encoded genes, such as DYRK1A, that lead to increased phosphorylation of tau directly and indirectly," Wolvetang explains.
"DYRK1A has also been implicated in AD."
While the researchers are careful to emphasise the limitations of their research – which is based on cultures of human cells in isolation, which can't serve as a comprehensive facsimile of actual Alzheimer's in human patients – they're nonetheless hopeful their methods could one day pave the way for future treatments.
"The research highlights that human stem cell-based disease modelling in the dish can provide new insights into the molecular mechanisms that conspire to cause Alzheimer's disease", Wolvetang says in a statement, "and, excitingly, this now opens the way for drug screening."
The findings are reported in Stem Cell Reports.